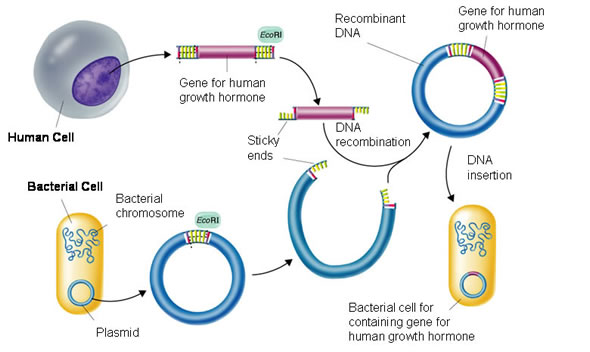
I. Mở đầu
Vào năm 1973, một nhóm các nhà khoa học đã tạo ra cơ thể sinh vật đầu tiên với các phân tử DNA tái tổ hợp. Theo đó, Cohen (ĐH Stanford, Mỹ) và Boyer (ĐH California, Mỹ) cùng các cộng sự đã đưa được một đoạn DNA từ một plasmid này vào một plasmid khác, tạo ra một plasmid hoàn toàn mới, plasmid tái tổ hợp. Sau đó, họ đưa plasmid tái tổ hợp vào trong các tế bào E. coli. Trong một thời gian ngắn, các tác giả này đã dùng các phương pháp giống nhau để gắn các gen từ hai loại vi khuẩn khác nhau, cũng như để chuyển các gen từ ếch vào vi khuẩn. Các thí nghiệm này đánh dấu một cuộc cách mạng vô cùng quan trọng trong lịch sử nghiên cứu khoa học của nhân loại.
Công nghệ DNA tái tổ hợp là một tập hợp các kỹ thuật phân tử để định vị, phân lập, biến đổi và nghiên cứu các đoạn DNA. Thuật ngữ tái tổ hợp được dùng thường xuyên do mục tiêu của nó là phối hợp DNA từ hai nguồn xa nhau. Ví dụ: các gen từ hai nguồn vi khuẩn khác nhau có thể được liên kết lại, hoặc một gen người có thể được đưa vào nhiễm sắc thể vi khuẩn. Công nghệ DNA tái tổ hợp (còn gọi là công nghệ di truyền, công nghệ gen hay kỹ thuật gen…) hiện nay bao gồm một mạng lưới các kỹ thuật phân tử được dùng để phân tích, biến đổi và tái tổ hợp hầu như mọi trình tự DNA.
1. Tác động của công nghệ DNA tái tổ hợp
Công nghệ DNA tái tổ hợp đã biến đổi sâu sắc phương thức nghiên cứu gen. Trước đây, thông tin về cấu trúc và tổ chức của gen thu được bằng cách kiểm tra biểu hiện kiểu hình của chúng, nhưng những kỹ thuật mới đã tạo ra khả năng tự đọc các trình tự nucleotide. Trước đây, các nhà di truyền phải chờ đợi sự xuất hiện các đột biến ngẫu nhiên hoặc cảm ứng để phân tích hiệu quả của sự sai khác di truyền, ngày nay họ có thể tạo ra đột biến ở các điểm nhất định một cách chính xác và xem chúng thay đổi kiểu hình như thế nào.
Công nghệ DNA tái tổ hợp đã cung cấp các thông tin mới về cấu trúc và chức năng của gen và đã thay đổi nhiều khái niệm cơ bản của di truyền học. Ví dụ: trong khi mã di truyền được xem là rất phổ biến, thì bây giờ chúng ta còn biết rằng các mã không phổ biến cũng tồn tại trong DNA ty thể. Trước đây, chúng ta nghĩ rằng tổ chức của các gen eukaryote giống với prokaryote, nhưng bây giờ chúng ta biết rằng nhiều gen eukaryote bị gián đoạn bởi các intron. Ngày nay, chúng ta đã biết đầy đủ hơn về các quá trình tái bản, phiên mã, dịch mã, biến đổi RNA (RNA processing) và điều hòa gen thông qua việc sử dụng các kỹ thuật tái tổ hợp DNA. Các kỹ thuật này cũng được dùng trong nhiều trong nhiều lĩnh vực khác, bao gồm hóa sinh học, vi sinh vật học, sinh học phát triển, sinh học thần kinh, tiến hóa và sinh thái học.
Công nghệ DNA tái tổ hợp cũng được ứng dụng để tạo ra nhiều sản phẩm thương mại, chẳng hạn: thuốc, hormone, enzyme và các giống cây trồng-vật nuôi. Một nền công nghiệp hoàn toàn mới, công nghiệp công nghệ sinh học, đã phát triển chung quanh việc sử dụng các kỹ thuật này để tạo ra các sản phẩm mới. Trong y học, các kỹ thuật tái tổ hợp DNA được dùng để thăm dò bản chất của ung thư, chẩn đoán các bệnh di truyền và nhiễm trùng, sản xuất thuốc và điều trị các rối loạn di truyền.
2. Làm việc ở mức độ phân tử
Kỹ thuật gen cho thấy một loạt cơ hội, mở ra các phương thức cần thiết (mà trước đây có thể không được) gần như là hiển nhiên. Vấn đề cơ bản đó là các gen có kích thước quá nhỏ và có hàng ngàn gen ở trong mỗi tế bào. Thậm chí, khi quan sát trên kính hiển vi mạnh nhất, thì DNA xuất hiện như là một sợi dây bé xíu, các nucleotide riêng rẽ không thể thấy, và không có một dấu hiệu nào về các đường nét vật lý ở chỗ bắt đầu và kết thúc của một gen.
Để minh họa vấn đề này, chúng ta hãy xem xét một ví dụ đặc trưng về di truyền phân tử như sau: Giả thiết rằng chúng ta muốn phân lập một gen đặc biệt của người và đặt nó vào trong vi khuẩn để sản xuất một lượng lớn các protein người đã được mã hóa. Vấn đề đầu tiên là tìm được gen mong muốn. Genome đơn bội của người chứa khoảng 3,3 tỷ cặp base của DNA. Giả sử gen mà chúng ta muốn phân lập dài 3.000 bp. Như vậy, gen đích của chúng ta chỉ chiếm một phần triệu của genome; vì thế để tìm kiếm gen của chúng ta trong một genome đồ sộ như thế là khó khăn hơn rất nhiều so với việc tìm kiếm một cây kim trong một đống cỏ khô. Nhưng thậm chí, nếu chúng ta có thể định vị gen, thì chúng ta sẽ tách nó ra khỏi genome như thế nào? Không có forcept đủ nhỏ để gắp một mảnh DNA đơn, và cũng không có một cái kéo cơ học nào đủ nhỏ để cắt ra khỏi genome một đoạn gen riêng biệt.
Nếu chúng ta thành công trong việc định vị và phân lập gen mong muốn, thì bước tiếp theo chúng ta cần đưa nó vào trong tế bào vi khuẩn. Các đoạn DNA mạch thẳng sẽ bị thoái biến nhanh bởi vi khuẩn; vì thế gen phải được chèn vào trong một dạng ổn định. Nó cũng phải ổn định để tái bản thành công hoặc nó sẽ không được phân chia tiếp khi tế bào phân chia.
Nếu chúng ta chuyển gen vào vi khuẩn thành công trong một dạng ổn định, chúng ta vẫn còn phải đảm bảo rằng gen được phiên mã và dịch mã. Sự biểu hiện của gen là một quá trình phức tạp đòi hỏi một số các trình tự DNA khác nằm ở bên ngoài gen. Tất cả những trình tự này phải hiện diện trong các hướng ở các vị trí thích hợp của chúng để sản xuất protein.
Cuối cùng, các phương pháp được sử dụng để phân lập và chuyển gen có hiệu quả vô cùng thấp, trong hàng triệu tế bào được hướng tới cho các phương thức này, chỉ có một tế bào có thể chọn lọc thành công và biểu hiện gen của người. Vì thế, chúng ta phải tìm kiếm nhiều tế bào vi khuẩn để phát hiện được một tế bào mang DNA tái tổ hợp.
Trước đây, các vấn đề này dường như là không vượt qua được. Nhưng ngày nay, các kỹ thuật phân tử được phát triển để khắc phục chúng, và các gen người được chuyển dễ dàng vào các tế bào vi khuẩn và ở đó chúng sẽ được biểu hiện tốt.
II. Endonuclease hạn chế
Trong tự nhiên, các enzyme endonuclease hạn chế (restriction endonuclease, RE), gọi tắt là enzyme
hạn chế, hiện diện trong hầu hết các tế bào vi khuẩn để ngăn cản DNA ngoại lai tiếp quản bộ máy tổng hợp protein của tế bào. DNA của chính chúng sẽ được bảo vệ khỏi tác dụng của enzyme hạn chế nhờ sự có mặt của các enzyme nội bào có thể methyl hóa (methylation) các nucleotide đặc biệt, vì thế các nucleotide này không được nhận biết bởi các enzyme hạn chế.
Việc phát hiện ra các enzyme hạn chế của vi khuẩn cắt DNA ở những trình tự đặc biệt, đã giúp cho việc thao tác gen dễ dàng hơn, do nó có thể giảm chiều dài của các phân tử DNA thành một tập hợp bao gồm các đoạn ngắn hơn. Hiện nay, các enzyme hạn chế được phân lập từ các sinh vật prokaryote.
Mỗi enzyme hạn chế chỉ nhận biết và cắt một trình tự DNA đặc biệt thường chứa bốn hoặc sáu cặp nucleotide. Ví dụ enzyme EcoRI tách chiết từ E. coli cắt trình tự GAATTC, enzyme BalI của Brevibacterium albidum cắt trình tự TGGCCA. Có hơn 900 enzyme hạn chế khác nhau được tinh sạch từ khoảng 250 chủng vi sinh vật. Các enzyme hạn chế cắt các phân tử DNA sợi đôi theo hai cách khác nhau:
- Cắt trên một đường thẳng đối xứng để tạo ra các phân tử đầu bằng (đầu thô).
- Cắt trên những vị trí nằm đối xứng quanh một đường thẳng đối xứng để tạo ra những phân tử đầu so le (đầu dính).
Vì một enzyme hạn chế chỉ nhận biết một trình tự duy nhất, cho nên số vị trí cắt trên một phân tử DNA đặc biệt thường là nhỏ. Các đoạn DNA được cắt bởi enzyme hạn chế có thể được phân tách theo kích thước bằng điện di agarose gel để nghiên cứu. Do sự tương tự của tổ chức phân tử trong tất cả các cơ thể, cho nên DNA vi khuẩn, DNA thực vật và DNA động vật có vú tương hợp nhau về cấu trúc. Vì thế, một đoạn DNA từ một dạng sống này có thể dễ dàng được pha trộn với DNA của một dạng sống khác. Sự tương tự này cũng phù hợp đối với plasmid, nhân tố di truyền ngoài nhân được tìm thấy trong nhiều loài vi khuẩn khác nhau. Chúng là những phân tử DNA mạch vòng đóng sợi đôi được dùng làm vector mang các đoạn DNA ngoại lai dùng trong kỹ thuật tái tổ hợp DNA. Giống như EcoRI, nhiều enzyme hạn chế đã tạo ra các đoạn DNA với đầu 5’ lồi (protruding). Một số enzyme hạn chế (ví dụ: PstI) đã tạo ra các đoạn DNA có đầu 3’ lồi. Một số enzyme hạn chế khác (ví dụ: BalI) cắt ở trục đối xứng để tạo ra các đoạn DNA mang đầu bằng (blunt) (Bảng 9.1).
Bảng 9.1. Trình tự nhận biết của một số enzyme hạn chế
1. Gắn các đầu bị cắt bởi enzyme hạn chế
Có hai kiểu gắn khác nhau: gắn đầu bằng và gắn đầu dính, bằng cách dùng enzyme DNA ligase của bacteriophage T4 có thể gắn các đầu bằng hoặc đầu dính lại với nhau, tuy nhiên trường hợp đầu bằng thường cho hiệu quả thấp hơn đầu dính.
Ví dụ: Hình 9.2 minh họa việc gắn các đầu dính được cắt bằng enzyme HindIII.
2. Isochizomer
Nói chung, các RE khác nhau nhận biết các trình tự khác nhau (Bảng 9.1), tuy nhiên cũng có một số trường hợp cho thấy có những enzyme được phân lập từ nhiều nguồn khác nhau nhưng lại cắt trong một trình tự, các enzyme đó được gọi là isochizomer. Hơn nữa, một vài enzyme nhận biết các chuỗi tetranucleotide mà trong một số trường hợp các tetranucleotide này lại xuất hiện trong các chuỗi hexanucleotide của các enzyme khác.
Ví dụ:
- MboI và Sau3A nhận biết trình tự:
- Trong khi đó BamHI nhận biết trình tự:
Trong một vài trường hợp các đoạn được tạo ra nhờ một enzyme hạn chế này khi gắn với các đoạn được tạo ra nhờ một enzyme hạn chế khác đã hình thành các thể lai mà các enzyme bố mẹ không thể nhận biết được. Ví dụ: SalI cắt trình tự (G↓TCGAC) và XhoI cắt trình tự (C↓TCGAG), các trình tự được sinh ra này đã gắn với nhau tạo thành thể lai (hybrid) mà các vị trí cắt hạn chế của chúng không thể nhận biết bởi các enzyme SalI và XhoI:
III. Phương thức tạo dòng
Các phương thức cơ bản của kỹ thuật DNA tái tổ hợp là: (1) Gắn một đoạn DNA vào một phân tử DNA (như là vector) có thể tái bản, và (2) cung cấp một môi trường cho phép sao chép phân tử DNA đã được gắn.
Có ba nhóm vector được dùng phổ biến để tạo dòng các đoạn DNA ngoại lai và tái bản (sao chép) trong E. coli; đó là plasmid, bacteriophage λ và cosmid. Tất cả những vector này phải có một số tính chất cần thiết sau:
- Chúng có khả năng tự tái bản trong E. coli.
- Mang các gen chỉ thị chọn lọc để dễ dàng phân biệt và tinh sạch vector của thể tái tổ hợp với các dạng khác.
- Chúng có các vùng DNA không cần thiết cho sự sinh sản trong vi khuẩn, vì thế DNA ngoại lai có thể được đưa vào trong các vùng này.
- Chúng có thể biến nạp vào tế bào vật chủ một cách dễ dàng.
1. Plasmid vector
Plasmid là các thông tin di truyền ngoài nhân có trong nhiều loài vi khuẩn khác nhau. Chúng là những phân tử DNA mạch vòng, sợi đôi, kích thước từ 1- ≥200 kb, có khả năng tự sao chép để tồn tại độc lập trong tế bào. Các plasmid có thể mang một số gen của vi khuẩn và các gen này có thể biểu hiện ra protein.
DNA của plasmid có thể được phân lập từ nuôi cấy vi khuẩn chứa plasmid bằng cách bổ sung chất tẩy (như là sodium dodecyl sulfate-SDS) và ly tâm sự sinh tan (lysate)1. Phức hợp nhiễm sắc thể vi khuẩn, lớn hơn plasmid nhiều, sẽ lắng xuống đáy của tube ly tâm, plasmid siêu xoắn và các đoạn nhiễm sắc thể mạch thẳng giữ lại trong thể nổi. Plasmid siêu xoắn một lần nữa được phân tách bằng ly tâm sau khi xử lý với CsCl và EtBr.
Plasmid mang các gen mã hóa cho các enzyme thường có lợi cho vi khuẩn vật chủ. Các plasmid có thể mang các kiểu hình khác nhau như: kháng kháng sinh, sản xuất kháng sinh, phân hủy các hợp chất hữu cơ phức tạp, sản xuất các enzyme hạn chế và enzyme biến đổi (modification enzymes).
Các plasmid có thể được chuyển vào trong vi khuẩn sau khi vi khuẩn được xử lý để tế bào có thể cho thấm qua nhất thời đối với các phân tử DNA nhỏ. Quá trình này được biết như là sự biến nạp (transformation). Vi khuẩn được biến nạp thành công có thể được chọn lọc dựa trên kiểu hình mới mà chúng nhận được từ plasmid, chẳng hạn khả năng kháng các kháng sinh.
Một số plasmid hiện diện trong tế bào có số bản sao thấp, một hoặc một vài bản sao trên tế bào, do DNA của plasmid chỉ sao chép một hoặc hai lần trước khi tế bào phân chia. Tuy nhiên, các plasmid khác tồn tại một số bản sao lớn hơn (10 tới 100 bản sao trên một tế bào) do DNA tái bản lặp lại cho đến khi đạt được số bản sao thích hợp. Các plasmid có số bản sao lớn được gọi là plasmid dạng xoắn lỏng lẻo (relaxed plasmid), và đây là một trong những tính chất hữu ích của vector tạo dòng.
Hình 9.3 trình bày một trong các plasmid vector thế hệ thứ hai dạng xoắn lỏng lẻo, pBR322, dài 4.363 bp, vector này chứa hai gen kháng kháng sinh là ampicillin (Amp) và tetracycline (Tet). Số thứ tự của các nucleotide trên vector được bắt đầu với vị trí EcoRI đơn: T đầu tiên trong chuỗi GAATTC được quy ước là nucleotide thứ nhất. Các số thứ tự sau đó được tiếp tục quanh phân tử vector theo hướng từ gen kháng tetracycline tới gen kháng ampicillin.
Hình 9.3. Plasmid vector pBR322. Apr (hay Ampr) và Tetr: gen kháng ampicillin và tetracycline, ori: trình tự khởi đầu sao chép, và một số vị trí nhận biết cho các RE.
Hình 9.4. trình bày một loại plasmid vector thế hệ thứ ba là pUC19, đây là loại vector tạo dòng đặc trưng, Nó mang vùng tạo dòng (multiple cloning sites) hay còn gọi là vùng đa nối (polylinker), vùng khởi đầu sao chép (ori), và hai gen chỉ thị (gen kháng ampicillin và gen lacZ’). Ampicillin là loại kháng sinh giết chết tế bào vi khuẩn, nhưng những vi khuẩn nào chứa vector pUC19 sẽ kháng lại loại kháng sinh này. Gen lacZ’ mã hóa enzyme β-galactosidase, bình thường enzyme này cắt lactose để sản xuất ra glucose và galactose. Enzyme này cũng cắt X-gal để tạo ra một cơ chất màu xanh; khi X-gal được bổ sung vào môi trường, các khuẩn lạc vi khuẩn chứa pUC19 sẽ có màu xanh và dễ dàng nhận biết. Vùng polylinker của vector pUC19 là tập hợp một số vị trí nhận biết đơn của các enzyme hạn chế cho phép gắn đoạn DNA ngoại lai vào plasmid.

Hình 9.4. Plasmid vector tạo dòng đặc trưng pUC19. Mang các vị trí cắt hạn chế đơn trong vùng tạo dòng, vùng khởi đầu sao chép (ori), và hai gen chỉ thị (gen Apr và gen lacZ’).
Plasmid có thể được cắt ở một vị trí xác định bằng enzyme hạn chế. Vì thế, các đoạn được tạo ra có thể tạo vòng bằng cách kết hợp các đầu dính bổ sung. Hơn nữa, các đoạn được tạo ra bởi một enzyme đặc biệt hoạt động trên một phân tử DNA sẽ có đầu tương đồng (đầu dính) với đoạn được tạo ra bởi cùng một enzyme hoạt động trên một phân tử DNA khác. Vì thế, các đoạn từ hai phân tử DNA khác nhau, từ hai cơ thể khác nhau có thể kết hợp bằng các liên kết hydrogen thuận nghịch khi các đoạn này được trộn lại.
Nếu sự kết hợp được gắn lại sau khi bắt cặp, thì các đoạn được kết hợp cố định bền vững, sự kết hợp của các đoạn này được thực hiện nhờ enzyme DNA ligase (còn gọi là polynucleotide ligase) liên kết cộng hóa trị các phân tử tái tổ hợp bằng cách tạo ra liên kết phosphodiester giữa nhóm 5’-PO4 của polynucleotide này với nhóm 3’-OH của polynucleotide khác.
Hình 9.5. Phương thức cơ bản để tạo dòng gen trong vi khuẩn E. coli
Hình 9.5 trình bày toàn bộ phương thức sản xuất DNA tái tổ hợp (tạo dòng gen). Plasmid được cắt ở các vị trí xác định bằng enzyme hạn chế. DNA của một genome ngoại lai được cắt bởi cùng một loại enzyme, một số đoạn trong đó có thể có gen quan tâm. Plasmid và các đoạn của genome được phối trộn và kết hợp nhờ enzyme DNA ligase. Các plasmid tái tổ hợp được biến nạp vào vi khuẩn bằng đồng nuôi cấy plasmid và vi khuẩn.
2. Bacteriophage λ vector
Các bacteriophage có nhiều ưu điểm khi được sử dụng làm vector tạo dòng. Vector dược sử dụng rộng rãi nhất là bacteriophage để gây nhiễm vào tế bào E. coli. Một trong những ưu điểm chính của phage λ là có hiệu quả chuyển DNA vào trong tế bào vi khuẩn cao. Ưu điểm thứ hai là một phần ba của genome phage λ là không cần thiết cho sự xâm nhiễm và sinh sản của nó trong tế bào vật chủ, không có những gen này, một tiểu thể λ vẫn chuyển thành công DNA của nó vào vi khuẩn và sinh sản được. Các gen không cần thiết này, khoảng >15 kb, có thể được thay thế bằng một đoạn DNA ngoại lai khoảng 1523 kb. Ưu điểm thứ ba là DNA sẽ không bị đóng gói trong vỏ λ trừ khi nó dài từ 40-50 kb; vì thế các đoạn DNA ngoại lai không bao giờ được chuyển vào tế bào trừ khi chúng được chèn vào trong genome phage λ, điều kiện cần thiết để đảm bảo đoạn DNA ngoại lai sẽ được tái bản sau khi nó vào trong tế bào vật chủ.
Các gen cần thiết của genome phage được định vị trong một cụm. Các chủng của phage λ, được gọi là vector thay thế, đã được biến đổi di truyền với một vị trí RE duy nhất cho EcoRI (chủng phage λ thế hệ mới EMBL 3 có ba vị trí cho các RE: EcoRI, BamHI và SalI) trên một mặt khác của các gen không cần thiết (Hình 9.6). Vì thế, có thể loại bỏ các gen không cần thiết bằng EcoRI. DNA ngoại lai được cắt bằng EcoRI sẽ có đầu dính bổ sung cho đầu của các đoạn DNA của phage λ cần thiết (nhánh trái và phải), để có thể được kết nối bằng DNA ligase. Genome của phage λ có các đoạn sợi đơn ngắn được gọi là các vị trí cos cần cho sự đóng gói DNA trong đầu của phage. Genome tái tổ hợp của phage λ sau đó có thể được đóng gói trong vỏ protein và được bổ sung vào trong E. coli. Các phage λ gây ra sự xâm nhiễm DNA tái tổ hợp của chúng vào trong tế bào vật chủ, nơi nó sẽ được tái bản. Chỉ các đoạn DNA có kích thước thích hợp và mang các gen cần thiết được đóng gói trong các vỏ của phage, cung cấp một hệ thống chọn lọc tự động cho các vector tái tổ hợp.

Hình 9.6. Bacteriophage λ là một vector tạo dòng hiệu quả
3. Cosmid vector
Các vector phage λ chỉ có thể mang các đoạn DNA có kích thước khoảng 15-23 kb. Tuy nhiên, các cosmid vector lại mang được các đoạn DNA có kích thước lớn hơn nhiều, khoảng 45 kb.
Hình 9.7. Tạo dòng trong cosmid. Hai vị trí cos gần vị trí cắt hạn chế ScaI và BamHI. DNA nguồn bị cắt bởi BamHI để phân đoạn có kích thước khoảng 45 kb. Phân tử plasmid DNA được cắt bởi BamHI và ScaI. Hai mẫu DNA này được trộn vào nhau và được gắn bằng T4 DNA ligase. Sau khi gắn xong, những phân tử này được đóng gói trong phần đầu của phage , và những phần tử có thể lây nhiễm sẽ được hình thành sau khi tạo đuôi.
Cosmid là các plasmid nhỏ mang các vị trí cos của phage; chúng có thể được đóng gói trong vỏ virus và được chuyển vào vi khuẩn nhờ sự xâm nhiễm của virus. Do tất cả các gen virus, ngoại trừ các vị trí cos, là không có, nên cosmid có thể mang các đoạn
DNA ngoại lai lớn hơn hai lần các đoạn mà phage vector có thể mang. Các cosmid vector có các thành phần sau: (1) một điểm khởi đầu sao chép của plasmid (ori); (2) một số các vị trí cắt hạn chế đơn; (3) một hoặc hơn các gen chỉ thị chọn lọc; và (4) các vị trí cos cho phép đóng gói DNA trong đầu của phage.
DNA ngoại lai được chèn vào trong cosmid trong cùng một phương thức với plasmid: cosmid và DNA ngoại lai đều được cắt bởi cùng một RE để tạo ra các đầu bổ sung (đầu dính), và chúng được liên kết với nhau bằng DNA ligase. Các cosmid tái tổ hợp được hợp nhất trong vỏ, và các tiểu thể phage được sử dụng để xâm nhiễm vào tế bào vi khuẩn, ở đó cosmid sẽ tái bản như một plasmid. Bảng 9.2 so sánh các tính chất của các vector plasmid, phage λ và cosmid.
Bảng 9.2. So sánh các vector plasmid, phage λ và cosmid
4. Thư viện cDNA
Thư viện cDNA (complementary DNA) là tập hợp các đoạn DNA bổ sung (cDNA) được tổng hợp từ mRNA của một bộ phận trong cơ thể sinh vật. Sử dụng thư viện cDNA có hai ưu điểm sau:
- Các dòng cDNA chứa trình tự mã hóa liên tục của một gen. Nhiều gen ở eukaryote là gián đoạn, có chứa nhiều đoạn intron. Sau khi cắt mRNA tiền thân (premRNA) và nối lại, các đoạn intron đã bị loại và mRNA hoàn chỉnh (mature mRNA) có trình tự mã hóa liên tục được tạo thành. Do cDNA được tạo thành từ khuôn mẫu mRNA hoàn chỉnh nên các dòng cDNA có thể tổng hợp protein cần thiết với số lượng lớn như mong muốn.
- Nhiều protein được tổng hợp với số lượng lớn do những tế bào chuyên hóa và như vậy trong các tế bào này mRNA của protein đó sẽ có tỷ lệ cao và thư viện cDNA được tạo ra từ các tế bào này sẽ có nhiều cDNA mã hóa cho các protein tương ứng. Sự dồi dào cDNA của một vài loại nào đó đã làm giảm nhẹ đáng kể việc xác định đúng dòng mong muốn từ thư viện gen.
Phương pháp tổng hợp gen từ mRNA ngày càng được phát triển, nó kết hợp với các phương pháp khác của sinh học phân tử cho phép ứng dụng trong nhiều lĩnh vực.
Xây dựng một thư viện cDNA bao gồm năm bước chính sau:
- Tinh sạch mRNA từ RNA tổng số của một phận cơ thể sinh vật.
- Tổng hợp sợi cDNA thứ nhất từ khuôn mẫu mRNA nhờ enzyme phiên mã ngược
(reverse transcriptase).
- Gây biến tính (đun sôi) thể lai mRNA-cDNA để phá hủy sợi mRNA bằng RNase H của E. coli. Tổng hợp sợi cDNA thứ hai từ khuôn mẫu sợi cDNA thứ nhất nhờ enzyme DNA polymerase với primer là vòng cặp tóc của nó để thu được phân tử cDNA sợi đôi.
- Cắt vòng cặp tóc bằng enzyme nuclease S1 và dùng enzyme Klenow sửa chữa hai đầu của sợi đôi cDNA để tạo ra đầu bằng.
- Gắn các đoạn nối (linker) vào hai đầu của cDNA sợi đôi trước khi tạo dòng trong vector thích hợp để xây dựng thư viện cDNA.
5. Thư viện genomic DNA
Thư viện genomic DNA là một tập hợp các đoạn DNA cùng đại diện cho một genome nguyên vẹn (hoặc gần nguyên vẹn) của cá thể mà DNA được bắt nguồn từ đó. Các thư viện được dùng chủ yếu hoặc để sàng lọc theo các phương pháp khác nhau nhằm phân lập một (hoặc các) trình tự nucleotide quan tâm đặc biệt, hoặc để xác định vị trí và thứ tự của các trình tự trong genome mà từ đó thư viện được xây dựng (physical mapping).
Xây dựng một thư viện genomic DNA bao gồm bốn bước chính: Cắt DNA, gắn DNA vào vector, đóng gói các dòng tái tổ hợp trong vỏ protein của virus để làm bước trung gian trước khi xâm nhập vào tế bào vật chủ, và chuyển cấu trúc đó vào tế bào vật chủ.
Thư viện genomic DNA có nhiều ứng dụng, chẳng hạn để lập bản đồ vật lý (physical mapping) của DNA và xác định các gen gây bệnh hoặc các chuỗi DNA quan tâm cho những phân tích khác.
Sản xuất ra các dòng mang các đoạn chèn DNA khác nhau nhưng gối lên nhau có nhiều thuận lợi và thư viện có thể được sử dụng trong quá trình chromosome walking. Chromosome walking thường được thực hiện với thư viện của cosmid, phage hoặc YAC2.
Các thư viện genomic DNA cũng cần thiết cho việc xác định các gen gây bệnh bằng cách tạo dòng chức năng (functional cloning). Theo hướng này, thông tin về chức năng của gen được khai thác để phân lập gen mong muốn từ thư viện. Một oligonucleotide có trình tự dựa trên chuỗi amino acid từng phần được dùng như là một probe (mẫu dò) để phân lập dòng cDNA bằng cách sàng lọc thư viện cDNA. Dòng cDNA này sau đó có thể được dùng để sàng lọc thư viện genome nhằm phân lập các dòng genomic DNA và cho phép khảo sát đặc điểm của chuỗi genomic hoàn chỉnh.
IV. Biểu hiện gen ngoại lai trong vi khuẩn
Về mặt lý thuyết, kỹ thuật DNA tái tổ hợp cho phép đưa bất kỳ một gen nào đó từ một sinh vật này vào một sinh vật khác. Vấn đề quan trọng là làm sao để gen ngoại lai có thể biểu hiện trong cơ thể vật chủ.
Để biểu hiện tất cả các gen ngoại lai trong E. coli phải bắt đầu bằng việc gắn đoạn gen ngoại lai vào trong vector biểu hiện (thường là plasmid). Các vector biểu hiện là vector có thể mang các gen ngoại lai mong muốn cho phép thực hiện sự phiên mã của các bản sao được tạo dòng và sự dịch mã các mRNA của chúng trong E. coli. Vector này phải có đủ các cấu trúc cần thiết sau:
- Các trình tự mã hóa gen chỉ thị (marker) để đảm bảo duy trì vector trong tế bào.
- Một promoter kiểm soát phiên mã (ví dụ: lac, trp hoặc tac) cho phép sản xuất một lượng lớn mRNA từ các gen được tạo dòng.
- Các trình tự kiểm soát dịch mã như vùng liên kết ribosome được bố trí thích hợp và codon khởi đầu AUG.
- Một polylinker để đưa gen ngoại lai vào trong một hướng chính xác với promoter.
Chỉ khi được cấu trúc đầy đủ như thế, các vector biểu hiện mang gen ngoại lai mới được biến nạp vào chủng E. coli thích hợp. Nếu đoạn gen ngoại lai không nằm giữa promoter và vị trí kết thúc phiên mã, thì nó sẽ không được phiên mã.
1. Các protein nguyên thể tái tổ hợp
Các protein nguyên thể (native protein) có thể được sản xuất trong E. coli bằng cách sử dụng promoter mạnh và một vùng liên kết ribosome (ribosome binding sitesRBS) hiệu quả. Để biểu hiện gen prokaryote có RBS mạnh chỉ cần cung cấp một promoter là đủ. Trong khi đó, để biểu hiện một gen eukaryote (hoặc một gen prokaryote với một RBS yếu) cần phải cung cấp cả promoter lẫn RBS.
1.1. Biểu hiện của gen prokaryote-Promoter
Bước đầu tiên khi biểu hiện các protein của eukaryote trong vi khuẩn là chọn một vector biểu hiện mang promoter mạnh của prokaryote. Các promoter thích hợp nhất cho biểu hiện của gen ngoại lai ở E. coli là những promoter có khả năng điều chỉnh mạnh. Điển hình là promoter (lai) trp-lac.
Promoter trp-lac còn gọi là promoter tac (một dạng promoter lai giữa promoter trp và promoter lac) đã được sử dụng thành công để sản xuất một lượng lớn protein trong E. coli (Hình 9.8). Promoter trp được điều chỉnh bởi gen ức chế trp và có thể được cảm ứng bởi sự bổ sung của 3-indolylacetic acid (3-IAA) vào môi trường hoặc bằng cách thiếu tryptophan. Trong khi đó, promoter lac được điều chỉnh bởi gen ức chế lac và vì thế có thể được cảm ứng nhờ bổ sung nhân tố cảm ứng isopropyl--D-thiogalactoside (IPTG) vào môi trường nuôi cấy vi khuẩn. Cuối cùng, promoter (lai) trp-lac mang đoạn trp-35 được gắn với đoạn lac-10 và operator lac được điều chỉnh bởi gen ức chế lac (lac repressor).
1.2. Biểu hiện của gen eukaryote-Promoter và vùng liên kết ribosome
Để có mức độ biểu hiện cao của gen ngoại lai trong E. coli, không những phải sử dụng các promoter mạnh để sản xuất một lượng lớn mRNA mà còn sử dụng các RBS để đảm bảo chắc chắn mRNA được dịch mã một cách có hiệu quả. Trong E. coli, RBS bao gồm một mã khởi đầu AUG và một trình tự nucleotide ngược hướng từ codon khởi đầu. Trình tự này được gọi là chuỗi Shine-Dalgarno (SD) giàu A-G, bổ sung cho đầu 3’ của 16S rRNA của E. coli. Liên kết của ribosome với mRNA được khởi đầu bằng cách bắt cặp giữa chuỗi SD trong mRNA và trình tự ở đầu 3’ của 16S rRNA.
Hình 9.8. Vector pKK177-3. pKK177-3 là một tac vector chứa vùng tạo dòng gen ngoại lai cùng hướng với promoter tac. Cùng hướng với vùng này là rrnB mang gen 5S của E. coli và hai nhân tố kết thúc phiên mã T1 và T2.
Hiệu suất dịch mã của mRNA chịu sự chi phối của một số yếu tố như sau :
- Mức độ bổ sung giữa chuỗi SD và đầu 3’ của 16S rRNA.
- Không gian và khả năng để chuỗi DNA nằm giữa chuỗi SD và codon AUG.
- Nucleotide theo sau AUG ảnh hưởng sự liên kết ribosome.
Đưa codon ATG vào trong gen được tạo dòng và duy trì RBS của vi khuẩn bằng cách dùng vector pAS1 (Hình 9.9). Vector pAS1 mang promoter PL và RBS của gen cII của bacteriophage , codon khởi đầu ATG được dung hợp trực tiếp với phần mã hóa đầu amino của gen eukaryote muốn biểu hiện. Cách làm này có thể hạn chế việc tối ưu khoảng cách giữa chuỗi SD của vi khuẩn và codon khởi đầu ATG của gen eukaryote để có được sự biểu hiện hiệu quả của gen. Đoạn DNA có gắn promoter và chuỗi SD sau đó được biến nạp vào các chủng E. coli thích hợp. Sàng lọc thể biến nạp để thu các khuẩn lạc có mức độ phiên mã cao.
Hình 9.9. Vector pAS1. Vector pAS1 là một plasmid dài khoảng 5,8 kb mang promoter PL của bacteriophage λ và vị trí cắt hạn chế duy nhất BamHI định vị ở codon khởi đầu ATG của gen cII của bacteriophage λ
.
2. Các protein dung hợp tái tổ hợp
2.1. Protein dung hợp
Protein dung hợp còn gọi là protein lai được mã hóa bởi một gen lai (fusion gene) do sự dung hợp in vitro các đoạn gen khác nhau. Vì vậy, protein dung hợp sẽ mang trình tự amino acid của hai protein khác biệt. Các protein dung hợp được xây dựng cho các mục đích khác nhau. Sự dung hợp gen được thực hiện bằng cách gắn phần mã hóa của gen được tạo dòng ở gần đầu tận cùng 3’ của gen lacZ. Protein dung hợp có những ưu điểm chính sau:
- Protein dung hợp thường được sản xuất với hàm lượng lớn do sự khởi đầu phiên mã và dịch mã được điều khiển bởi các trình tự tiêu chuẩn của E. coli.
- Dung hợp các trình tự ngoại lai với các gen của E. coli thường cho kết quả các sản phẩm ổn định hơn các protein ngoại lai nguyên thể.
2.2. Vector biểu hiện các gen dung hợp với gen lacZ
Một số hệ thống vector được phát triển để biểu hiện các gen dung hợp với gen lacZ, điển hình là các
vector họ pUR (Hình 9.10). Chọn vector và vị trí cắt hạn chế thích hợp người ta có thể tiến hành dung hợp cho hầu hết gen được tạo dòng.
2.3. Phát hiện các protein dung hợp
Gắn vector plasmid (ví dụ: pUR) thích hợp với đoạn DNA ngoại lai để tạo ra một sự dung hợp trong khung đọc. Biến nạp các vector này vào E. coli. Kiểm tra các khuẩn lạc riêng biệt mang đoạn DNA ngoại lai mong muốn bằng cách tách chiết DNA của vector plasmid, sau đó cắt bằng enzyme hạn chế thích hợp và điện di kiểm tra trên agarose gel. Sàng lọc các khuẩn lạc sản xuất protein dung hợp.
2.4. Tách chiết các protein dung hợp để sản xuất kháng thể
Protein dung hợp có thể được tách chiết theo một số cách: dùng dịch chiết urea, sắc ký ái lực aminophenylthiogalactoside, điện di sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) hoặc phối hợp giữa các cách này. Kỹ thuật đơn giản nhất là SDS-PAGE, nhuộm gel và phát hiện băng protein mới, sau đó cắt băng này ra khỏi gel, đông khô rồi nghiền thành bột mịn. Bột này sẽ được dùng để tiêm vào thỏ để sản xuất kháng thể.
Hình 9.10. Các vector họ pUR. Đây là các vector dung hợp với gen lacZ, có các vị trí tạo dòng
BamHI, SalI, PstI, XbaI, HindIII và ClaI ở đầu 3’ của gen lacZ. Sự chèn vào của đoạn DNA ngoại lai (cDNA) trong các vị trí tạo dòng thích hợp cho phép sản xuất protein dung hợp của -galactosidase hoạt động với chuỗi peptide được mã hóa bởi DNA ngoại lai.
3. Xác định mức độ biểu hiện của gen được tạo dòng
Nói chung, có ba cách thường được dùng để xác định mức độ biểu hiện protein ngoại lai của gen được tạo dòng:
- Điện di polyacrylamide gel có SDS để xác định protein có kích thước thích hợp được sản xuất ở mức độ cao trong các tế bào mang vector biểu hiện. Thông thường, protein quan tâm có thể quan sát bằng cách nhuộm gel với Coomassie Brilliant Blue hoặc bằng AgNO3. Nếu không thấy có băng protein mới khi dùng các thuốc nhuộm này, thì đánh dấu sự trao đổi chất với 100 µCi của [35S]Met hoặc [35S]Cys trên 1 mL dịch nuôi cấy trong 5 phút. Sau đó, sử dụng kỹ thuật SDS-PAGE hoặc phóng xạ tự ghi có thể phát hiện được protein quan tâm.
- Phân tích Western blot bằng cách dùng các kháng thể liên kết đặc hiệu với protein quan tâm đã được thẩm tích lên màng nitrocellulose sau khi thực hiện điện di SDS-
polyacrylamide gel.
- Nếu mức độ biểu hiện thấp thì nên đặt gen lacZ cùng hướng với gen được biểu hiện. Như vậy, nếu sự phiên mã hoặc dịch mã hạn chế biểu hiện thì những thay đổi trong hệ thống biểu hiện có thể được kiểm soát bằng những thay đổi trong hoạt tính của βgalactosidase.
▪ Phân tích Western blot
- Kỹ thuật SDS-PAGE
Là kỹ thuật điện di trên polyacrylamide gel với sự có mặt của SDS cho phép phân tách các phân tử protein có khối lượng khác nhau. SDS có điện tích âm rất lớn và có khả năng liên kết với mạch peptide. Như vậy, số lượng SDS tương tác với protein tỷ lệ với kích thước phân tử protein và điện tích của SDS bám vào có thể làm bất cứ phân tử protein nào cũng chuyển động trong điện trường từ cực âm sang cực dương. Do đó, bằng phương pháp điện di, có thể phân tách riêng biệt các phân tử protein có khối lượng phân tử khác nhau.
- Phản ứng lai kháng nguyên-kháng thể
Phản ứng kháng nguyên-kháng thể có tính đặc hiệu rất cao. Vì vậy, có thể áp dụng phản ứng này để phát hiện sự có mặt và tinh sạch protein. Kháng thể (antibody) được sản xuất khi đưa kháng nguyên vào động vật thí nghiệm (thỏ, chuột…) và được tinh sạch từ máu động vật sau khi gây nhiễm. Những kháng thể tạo ra bằng cách này là những kháng thể đa dòng (polyclonal antibodies-do các tế bào lympho khác nhau tiết ra), do đó chúng có khả năng nhận biết một số kháng nguyên. Ngược lại, kháng thể đơn dòng (monoclonal antibodies) chỉ tương tác với một kháng nguyên nhất định.
Kháng thể được đánh dấu bằng enzyme (hoặc bằng chất phát huỳnh quang) để phát hiện protein đặc hiệu (thường được thẩm tích/chuyển thấm lên màng nitrocellulose sau khi chạy điện di SDS, và cố định ở đó) thông qua kỹ thuật Western blot (hoặc immunoblot) (Hình 9.11). Sau khi protein trên màng lai gắn với kháng thể thứ nhất đặc hiệu và tiếp đến là kháng thể thứ hai có đánh dấu enzyme (alkaline phosphatase, horseradish peroxidase…) thì phức hợp này sẽ được liên kết với cơ chất để tạo màu. Sự hiện diện của protein ngoại lai (sản phẩm dịch mã của gen ngoại lai được chuyển vào tế bào vật chủ) sẽ được phát hiện nhờ sự xuất hiện màu của phản ứng lai.
Sự phân bố của protein đặc hiệu trong tế bào và tổ chức mô cũng có thể phát hiện bằng kỹ thuật lai in situ (in situ hybridization) với nguyên tắc tương tự Western blot. Ngoài ra, kháng thể được sử dụng để tinh sạch protein đặc hiệu bằng kết tủa miễn dịch hoặc sắc ký ái lực (affinity chromatography). Kháng thể đánh dấu được dùng để định lượng kháng nguyên trong kỹ thuật xét nghiệm hấp thụ miễn dịch liên kết enzyme (enzyme-linked immunosorbent assay) gọi tắt là ELISA (Hình 9.11).
V. Phương pháp phát hiện dòng vi khuẩn có DNA tái tổ hợp
1. Lai khuẩn lạc và vết tan
DNA được tạo dòng trong plasmid hoặc YAC sản xuất ra các khuẩn lạc khi các nuôi cấy biến nạp được dàn mỏng trên đĩa agar chứa môi trường sinh trưởng và nuôi cấy dưới những điều kiện thích hợp. Các bacteriophage sinh tan tế bào bị chúng xâm nhiễm và sản xuất ra các plaque (vết tan) có dạng hình tròn chu vi khoảng 2-3 mm có màu sáng trên thảm vi khuẩn (bacterial lawn) của lớp agar đỉnh (trong trường hợp này người ta chuẩn bị đĩa agar có hai lớp: lớp agar đỉnh có nồng độ agar thấp để bacteriophage dễ sinh tan tế bào vi khuẩn, và lớp agar đáy có nồng độ agar cao hơn).
Các vector, như mô tả ở trên, thường chứa gen chỉ thị cho phép chọn lọc những tế bào vật chủ mang vector (thể biến nạp). Các chỉ thị này thường là gen kháng kháng sinh và các tế bào biến nạp sinh trưởng trên môi trường chứa kháng sinh tương ứng.
Hình 9.11. Kỹ thuật Western blot và ELISA dựa trên phản ứng liên kết kháng nguyênkháng thể
Ngoài ra, các vector còn chứa các gen chỉ thị bổ sung để phân biệt các tế bào biến nạp chứa đoạn chèn của DNA ngoại lai với các tế bào chứa các vector tự tái tạo lại vòng. Ví dụ: gen lacZ mã hóa enzyme β-galactosidase.
Một dòng (clone) chứa các chuỗi DNA quan tâm đặc biệt có thể được xác định bởi lai khuẩn lạc hoặc vết tan. Một lượng nhỏ khuẩn lạc biến nạp hoặc vết tan được chuyển lên màng nitrocellulose hoặc nylon bằng cách phủ (overlay) màng này lên trên đĩa agar. DNA được biến tính và cố định trên màng bằng đun nóng (baking) hoặc chiếu tia cực tím (UV-crosslinking), và sau đó được lai trong đệm chứa probe đã đánh dấu đồng vị phóng xạ có trình tự bổ sung một phần của chuỗi được xác định. Ví dụ: Có thể một oligonucleotide tổng hợp nhân tạo, có nguồn gốc từ genomic DNA từng phần, cDNA hoặc trình tự protein hoặc sản phẩm PCR. Trong một vài trường hợp, probe có thể được thiết kế dựa trên trình tự bắt nguồn từ gen tương đồng của các loài khác và thường được lai với cường lực thấp. Các probe thừa được rửa khỏi màng để ủ với phim X-quang. Theo hướng của phim (sau khi rửa) so sánh với đĩa agar gốc, sau đó sẽ có khả năng gắn kết các khuẩn lạc thực tế với các khuẩn lạc lai dương tính tương ứng trên phim X-quang.
2. Sàng lọc thư viện gen bằng PCR
PCR (polymerase chain reaction) cũng có thể được ứng dụng để sàng lọc các thư viện genomic DNA hoặc cDNA được xây dựng trong các plasmid vector hoặc bacteriophage vector. Ưu điểm chính của sàng lọc bằng PCR so với sàng lọc dựa trên lai truyền thống là ít tốn thời gian hơn. Sàng lọc bằng PCR có thể được đảm bảo trong 3-4 giờ trong khi đó nó có thể mất một vài ngày trước khi phát hiện bằng kỹ thuật lai phân tử (molecular hybridization). Kỹ thuật sàng lọc bằng PCR thể hiện chỉ thị về kích thước của các đoạn chèn DNA được tạo dòng hơn là trình tự của đoạn chèn, tuy nhiên các primer PCR đặc trưng cho đoạn chèn DNA ngoại lai cũng có thể được sử dụng. Điều này cho phép khảo sát chính xác hơn đặc điểm của các dòng từ thư viện cDNA và genomic DNA.
▪ Nguyên tắc của PCR
PCR là một kỹ thuật được sử dụng phổ biến trong công nghệ sinh học hiện đại và đã đóng góp rất lớn cho những tiến bộ về sinh học phân tử, đánh dấu một bước tiến vô cùng quan trọng tương đương với việc khám phá ra các enzyme hạn chế và kỹ thuật Southern blot.
Kỹ thuật PCR dựa trên cơ sở phản ứng mở rộng primer nhờ enzyme Taq polymerase để khuếch đại in vitro các nucleic acid đặc trưng. PCR cho phép khuếch đại theo hàm mũ lên đến hàng triệu lần các đoạn DNA có chiều dài khoảng từ 200-3.000 bp. Đoạn DNA được khuếch đại (DNA đích) được nhận diện nhờ cặp primer đặc trưng (oligonucleotide) thường có chiều dài khoảng 20 nucleotide.
Taq polymerase là một loại enzyme DNA polymerase chịu nhiệt (có ở vi khuẩn chịu nhiệt độ cao Thermus aquaticus) được dùng để tổng hợp các đoạn DNA mới trong môi trường có 4 loại deoxyribonucleotide (dATP, dCTP, dGTP và dTTP) và hai primer, trên cơ sở khuôn mẫu của một đoạn DNA nhất định đã biết hoặc chưa biết trình tự. Các đoạn DNA mới hình thành lại được sử dụng làm khuôn mẫu. Sau nhiều chu kỳ, số lượng đoạn DNA nói trên được nhân lên gấp nhiều lần, nhờ vậy có thể đủ số lượng để tách ra, phân tích trình tự hoặc tạo dòng... Primer ở bên trái tác động trên sợi DNA 3’-5’ được gọi là primer thuận (forward primer-ký hiệu là F). Primer ở bên phải tác động trên sợi DNA 5’-3’ được gọi là primer ngược (reverse primer-ký hiệu là R).
Nguyên tắc của PCR được trình bày trong hình 9.12. Theo đó, từ chu kỳ thứ hai Taq polymerase bắt đầu tạo ra các đoạn DNA có chiều dài xác định. Các primer thường là một oligonucleotide tổng hợp (synthetic oligonucleotide) có khoảng 10-20 nucleotide hoặc dài hơn. Nếu biết trình tự của đoạn gen cần khuếch đại thì có thể tổng hợp nhân tạo các primer tương ứng để thực hiện PCR và tách chúng ra bằng kỹ thuật điện di. PCR thường tiến hành khoảng 25-35 chu kỳ, qua đó từ 10-6μg DNA ban đầu có thể khuếch đại (amplification) lên tới trên 1μg (khoảng 2 kb). Mỗi chu kỳ PCR bao gồm ba giai đoạn có nhiệt độ khác nhau:
- Gây biến tính (denaturation) ở 90-95oC. Trong giai đoạn biến tính phân tử DNA khuôn mẫu ở dạng xoắn kép được tách thành hai sợi đơn (single strands).
- Gắn primer (annealing) ở 40-65oC. Trong giai đoạn này các primer gắn vào các vị trí có trình tự tương đồng ở DNA khuôn mẫu. Các liên kết ion tạo thành một đoạn nhỏ (các primer đã lắp ráp chính xác) và trên các đoạn nhỏ DNA sợi đôi đó (khuôn mẫu và primer) enzyme Taq polymerase có thể bắt đầu quá trình sao chép khuôn mẫu.
- Kéo dài phân tử (extension) ở 70-72oC. Đây là khoảng nhiệt độ tối thích cho Taq polymerase tiến hành tổng hợp DNA bắt đầu từ các vị trí có primer theo chiều 5’→3’.
Hình 9.12. Sơ đồ phản ứng chuỗi DNA polymerase (PCR)
3. Sàng lọc các thư viện cDNA bằng các probe khác nhau
Sàng lọc thư viện cDNA bằng cách lai nucleic acid là phương pháp được sử dụng phổ biến và đáng tin cậy nhất để tìm kiếm các dòng quan tâm. Sàng lọc bằng phương pháp lai nucleic acid cho phép phân tích đồng thời nhiều dòng và nhanh, không đòi hỏi các dòng cDNA phải hoàn chỉnh và sản phẩm được tổng hợp trong tế bào vật chủ phải có hoạt tính sinh học hoặc kháng nguyên. Hơn nữa, cơ sở lý thuyết của kỹ thuật lai nucleic acid đã được hiểu biết đầy đủ. Điều này cho phép phát triển một số lượng lớn các kỹ thuật khác nhau có thể điều chỉnh các probe của nucleic acid có các chiều dài và đặc điểm khác nhau.
- Các probe tương đồng. Các probe tương đồng chứa ít nhất một phần của chuỗi nucleic acid chính xác của dòng cDNA quan tâm. Chúng được dùng trong nhiều trường hợp khác nhau, ví dụ: khi một dòng bộ phận của cDNA hiện có được sử dụng để phân lập một dòng hoàn chỉnh từ thư viện cDNA. Thông thường, đoạn bắt nguồn từ một đầu hoặc đầu kia của dòng hiện có được phân lập, đánh dấu phóng xạ in vitro và dùng để thăm dò thư viện. Lai với các probe tương đồng thường được tiến hành dưới các điều kiện nghiêm ngặt.
- Các probe tương đồng từng phần. Các probe tương đồng từng phần được dùng để phát hiện các dòng cDNA có quan hệ họ hàng, nhưng không giống hệt nhau hoàn toàn, với các trình tự của probe.
- Các probe oligonucleotide nhân tạo. Các probe oligonucleotide nhân tạo là các vùng dNTP của trình tự xác định được tổng hợp in vitro. Trình tự của các probe này được suy luận, bằng cách dùng mã di truyền, từ các vùng ngắn của chuỗi amino acid đã biết của protein quan tâm. Do sự thoái biến của mã di truyền3, chuỗi amino acid đề xuất có thể không được đặc trưng chính xác bởi các oligonucleotide đơn được dự đoán cho trình tự. Mặc dù, trong rất nhiều trường hợp, trình tự các amino acid giống nhau có thể được đặc trưng bởi nhiều oligonucleotide khác nhau.
4. Phân tích genomic DNA bằng phương pháp lai Southern
Việc phát hiện và xác định các chuỗi nucleic acid đặc trưng là công việc thường xuyên trong nghiên cứu sinh học phân tử. Nguyên tắc của kỹ thuật này là dựa vào sự lai phân tử, dưới các điều kiện thích hợp hai chuỗi nucleic acid đơn tạo thành một phân tử lai. Phản ứng lai phụ thuộc rất nhiều vào mức độ tương đồng của hai chuỗi nucleotide. Việc tạo thành phân tử sợi đôi như thế xảy ra chủ yếu thông qua liên kết hydrogen giữa các base guanosine với cytosine, và adenosine với thymidine. Thành phần và sự phân bố của các base không giống rõ rệt với các phân tử nucleic acid cho kết quả các tính chất lai khác nhau, vì thế liên kết hydrogen (hoặc lai phân tử giữa các base tương đồng) khi được ghép cặp thích hợp đã cung cấp một công cụ có giá trị để xác định các chuỗi liên quan hoặc đồng nhất với chuỗi nucleic acid quan tâm (probe).
Trong số các kỹ thuật khác nhau sử dụng lai phân tử để phân tích nucleic acid, thì kỹ thuật được sử dụng phổ biến nhất là lai mẫu dò nucleic acid được đánh dấu với nucleic acid đích được cố định trên một vật đỡ rắn, đặc trưng là màng nitrocellulose hoặc nylon.
Trình tự của kỹ thuật lai Southern blot bao gồm các bước sau: phân tách các đoạn cắt hạn chế của DNA hệ gen bằng điện di agarose gel, chuyển DNA từ agarose gel lên màng lai, lai các mẫu dò được đánh dấu đồng vị phóng xạ với các nucleic acid được cố định trên màng lai (Hình 9.13).
Hình 9.13. Sơ đồ của kỹ thuật lai Southern blot. Các mẫu DNA đích và genomic DNA (A) và (B) trong ví dụ này được cắt bằng EcoRI và được phân đoạn bằng điện di trên agarose gel. DNA của plasmid mang đoạn chèn DNA dùng làm mẫu dò (C) cũng được cắt bằng EcoRI và điện di như là một đối chứng dương tính.
Các thí nghiệm lai trên màng thường được tiến hành bằng cách đánh dấu probe bằng 32P (mặc dù các đồng vị phóng xạ khác như 35S cũng có thể được sử dụng). Phản ứng lai của Southern đòi hỏi hoạt tính đặc hiệu của probe tối thiểu phải là 109 dpm/μg, mặc dù hoạt tính 108 dpm/μg có thể được xem như là chấp nhận được trong các ứng dụng cần cường lực thấp. Các phương pháp không dùng đồng vị phóng xạ (ví dụ: digoxigenindUTP) ngày càng được sử dụng phổ biến hơn mặc dù độ nhạy của chúng là không lớn. Nhưng các kỹ thuật không dùng đồng vị phóng xạ an toàn hơn cho nghiên cứu viên, tạo ra các probe có thể được bảo quản trong thời gian dài trước khi dùng và kết quả phát hiện các tín hiệu lai nhanh hơn.
VI. Các ứng dụng của công nghệ DNA tái tổ hợp
Công nghệ DNA tái tổ hợp hiện nay có rất nhiều ứng dụng trong đời sống. Các ứng dụng này bao gồm sản xuất dược phẩm và các loại hóa chất khác, các vi khuẩn đặc biệt, các cây trồng nông nghiệp quan trọng, và các vật nuôi đã được biến đổi di truyền... Kỹ thuật này cũng được sử dụng rộng rãi trong các xét nghiệm y khoa và (trong một vài trường hợp) đang được dùng để kiểm tra các khiếm khuyết di truyền ở người. Hàng trăm công ty hiện nay đang đặc biệt phát triển các sản phẩm thông qua biến đổi di truyền các cơ thể sống. Dưới đây là các ứng dụng chính của công nghệ DNA tái tổ hợp.
1. Ứng dụng trong dược phẩm
Sản phẩm thương mại đầu tiên được phát triển bằng công nghệ DNA tái tổ hợp là các dược phẩm dùng trong điều trị các bệnh và các rối loạn ở người. Năm 1979, Tổ hợp Eli Lilly bắt đầu sản xuất thương mại insulin người bằng công nghệ DNA tái tổ hợp. Trước thời điểm này, tất cả insulin dùng trong điều trị bệnh tiểu đường được tách chiết từ tụy của các vật nuôi cho thịt. Mặc dù nguồn insulin này có tác dụng tốt cho nhiều người mắc bệnh tiểu đường, nhưng nó vẫn không phải là insulin người, và một số người đã trải qua các phản ứng phụ với protein ngoại lai. Gen insulin người sau đó đã được chèn vào plasmid và chuyển vào vi khuẩn để sản xuất insulin người. Các dược phẩm được sản xuất thông qua công nghệ DNA tái tổ hợp bao gồm: hormone sinh trưởng người (cho trẻ em mắc bệnh còi), nhân tố tạo tơ huyết (chữa bệnh khó đông máu), hoạt tố plasminogen mô (dùng để hòa tan các huyết khối trong bệnh nhân bị nhồi máu cơ tim), interferon, interleukin, DNA vaccine…
2. Các vi khuẩn đặc biệt
Các vi khuẩn có vai trò quan trọng trong các quá trình công nghiệp, bao gồm sản xuất ethanol từ các nguyên liệu thực vật, lọc khoáng từ quặng, xử lý nước thải và các loại chất thải khác. Các vi khuẩn đang sử dụng trong các quá trình này được biến đổi di truyền bằng công nghệ DNA tái tổ hợp sao cho chúng có thể hoạt động hiệu quả hơn. Các chủng vi khuẩn mới hữu ích thu được từ kỹ thuật hiện đại này đang được phát triển để phân hủy các hóa chất độc và các chất gây ô nhiễm, tăng cường thu hồi dầu, tăng nitrogen cho cây trồng, và ức chế sinh trưởng của các vi khuẩn và vi nấm gây bệnh.
3. Các sản phẩm nông nghiệp
Công nghệ DNA tái tổ hợp cũng đã có một ảnh hưởng quan trọng đến sản xuất nông nghiệp, nơi mà hiện nay nó được dùng để tạo ra các cây trồng và vật nuôi mang các đặc điểm có giá trị. Trong nhiều năm, các nhà bệnh học thực vật đã thừa nhận rằng thực vật bị nhiễm bệnh virus thể nhẹ sau đó sẽ kháng lại sự nhiễm trùng các chủng mang độc tính mạnh. Vì vậy, các nhà di truyền học đã tạo ra tính kháng virus trong cây trồng bằng cách chuyển các gen vỏ protein của virus vào tế bào thực vật. Cây bí (squash) biến đổi di truyền có tên là Freedom II, mang các gen của virus 2 gây bệnh khảm ở dưa hấu (watermelon mosaic virus 2) và virus gây bệnh khảm màu vàng ở cây zucchini (zucchini yellow mosaic virus) để chống sự nhiễm trùng virus.
Một hướng khác được biến đổi di truyền đó là tính kháng sâu (pest) ở thực vật đã làm giảm sự phụ thuộc vào các thuốc trừ sâu hóa học. Độc tố protein từ vi khuẩn Bacillus thuringiensis đã giết một cách chọn lọc các ấu trùng của các sâu bọ côn trùng nhất định nhưng lại không gây độc cho sự sống của các động vật hoang dã, người và các loài côn trùng khác. Gen độc tố được phân lập từ vi khuẩn, liên kết với promoter hoạt động sau đó được chuyển vào trong ngô, cà chua, khoai tây và bông. Gen này đã sản xuất độc tố đối với côn trùng trong cây, và sâu bướm khi ăn cây đã bị chết.
Công nghệ DNA tái tổ hợp cũng đã cho phép phát triển tính kháng chất diệt cỏ ở thực vật. Một vấn đề quan trọng trong nông nghiệp là điều khiển các cỏ dại cạnh tranh nước, ánh sáng và chất dinh dưỡng với cây trồng. Mặc dù chất diệt cỏ có tác dụng giết cỏ dại, nhưng chúng cũng có thể gây nguy hiểm cho cây trồng. Các gen cung cấp tính kháng cho nhiều loại chất cỏ dại đã được chuyển vào cà chua, đậu tương, bông, cây cải dầu và các cây trồng thương mại quan trọng khác. Khi đồng ruộng có canh tác các cây trồng này được phun thuốc diệt cỏ, thì cỏ dại bị giết nhưng các cây trồng biến đổi di truyền lại không bị ảnh hưởng. Năm 1999, đã có hơn 21 triệu ha cây đậu tương biến đổi di truyền và 11 triệu ha cây ngô chuyển gen được trồng trên thế giới.
Công nghệ DNA tái tổ hợp cũng được ứng dụng cho các vật nuôi. Ví dụ: gen sản xuất hormone sinh trưởng được phân lập từ gia súc và được tạo dòng trong E. coli, các vi khuẩn này sản xuất một lượng lớn hormone sinh trưởng của bò, yếu tố liên quan đến tăng sản xuất sữa. Các động vật chuyển gen được phát triển để mang gen mã hóa cho các sản phẩm dược liệu. Ví dụ: một gen người mã hóa cho nhân tố VIII (hình thành tơ huyết) được liên kết với vùng điều hòa của gen sản xuất β-lactoglobulin ở cừu, một protein tạo sữa. Gen dung hợp được tiêm vào phôi cừu, tạo ra một con cừu chuyển gen có khả năng sản xuất trong sữa của nó nhân tố đông máu của người. Một phương thức tương tự được dùng để chuyển gen α1-antitrysin, một protein dùng để điều trị bệnh nhân khí thũng di truyền (hereditary emphysema), vào trong cừu. Cừu cái mang gen này sản xuất rất nhiều α 1-antitrysin khoảng 15g trong 1 lít sữa của chúng.
4. Thuốc oligonucleotide
Một ứng dụng công nghệ DNA tái tổ hợp trong thời gian gần đây đó là phát triển các thuốc oligonucleotide có trình tự phân tử DNA hoặc RNA nhân tạo ngắn có thể được dùng để điều trị bệnh. Các antisense oligonucleotide bổ sung cho các RNA không mong muốn, như là RNA của virus. Khi được bổ sung vào tế bào, thì các antisense DNA này liên kết với mRNA của virus và ức chế sự dịch mã của chúng.
Các DNA oligonucleotide sợi đơn liên kết chặt chẽ với các trình tự DNA khác, tạo thành một phân tử triplex DNA. Sự tạo thành triplex DNA cản trở liên kết của RNA polymerase và các protein khác cần cho sự phiên mã. Các oligonucleotide khác là các ribozyme, các phân tử RNA mà chức năng như là các enzyme. Những phức hợp này liên kết với các phân tử mRNA đặc hiệu và cắt chúng thành từng đoạn, phá hủy khả năng mã hóa protein của chúng. Một số thuốc oligonucleotide đã sẵn sàng để thử nghiệm trong điều trị bệnh AIDS và ung thư.
5. Liệu pháp gen
Đối với loại bệnh di truyền, bệnh do đột biến gen người ta phải cần đến sự can thiệp của liệu pháp gen, một hướng chữa bệnh gắn liền với các kỹ thuật tiên tiến trong lĩnh vực công nghệ sinh học hiện đại như các vi thao tác gen, sửa đổi thay thế gen...
Liệu pháp gen (gene therapy) thực chất là phương pháp chữa bệnh bằng gen. Có nhiều khái niệm khác nhau về liệu pháp gen, nhưng cách hiểu chung nhất là tập hợp các biện pháp để sử dụng các gen cần thiết (còn gọi là gen trị liệu) nhằm mục đích chữa bệnh cho con người.
Trong đó, gen trị liệu có thể là:
- Các gen hoạt động bình thường (gen lành) có thể đưa vào tế bào để thay thế gen hỏng, gen mất chức năng, khôi phục hoạt động bình thường của tế bào và sự sống của cơ thể.
- Là những gen có khả năng mã hóa một protein đặc hiệu, khi đưa vào tế bào sống có thể tạo nên các loại protein đặc hiệu. Các loại protein đặc hiệu có thể ức chế hoạt động của một gen khác trong tế bào, kìm hãm khả năng phân chia của tế bào hoặc gây chết các tế bào bị bệnh.
- Những gen khi đưa vào tế bào hoạt động đồng thời với các gen bệnh (gen bị đột biến trong tế bào) làm hạn chế tác động của gen bệnh hoặc hỗ trợ, bù đắp cho các gen bị hỏng.
- Gen trị liệu còn là các gen bất hoạt được đưa vào tế bào thay thế cho một gen lành nào đó, nhằm hạn chế sản phẩm không cần thiết của gen lành hoặc tạo ra cho tế bào một trạng thái mới, có tác dụng chống lại bệnh tật.
- Gen trị liệu có thể là những đoạn oligonucleotide có tác dụng kìm hãm hoạt động của gen bị hỏng, bị đột biến trong tế bào.
6. Chẩn đoán bệnh để can thiệp sớm
Bằng kỹ thuật chọc ối, kết hợp đồng thời phân tích máu bố mẹ bằng enzyme hạn chế, người ta có thể chẩn đoán sớm trước khi sinh (vì enzyme hạn chế có khả năng phân biệt được gen đột biến với gen bình thường). Các đoạn DNA cắt ra, được phân tách qua phương pháp điện di, đem lai với các mẫu dò DNA hoặc RNA đã đánh dấu bằng đồng vị phóng xạ 32P hoặc huỳnh quang. Ảnh phóng xạ tự ghi hoặc tín hiệu huỳnh quang cho ta thấy các đoạn DNA được lai với các mẫu dò. Các đoạn này được tách ra để nghiên cứu và xác định đột biến hay bình thường bằng enzyme hạn chế, vì một số đột biến di truyền có ảnh hưởng đến các vị trí dành cho enzyme hạn chế.
Ứng dụng các kỹ thuật sinh học phân tử trong lĩnh vực này có nhiều thuận lợi hơn các phương pháp kinh điển. Thứ nhất, có thể rút ngắn được thời gian nhờ ứng dụng kỹ thuật PCR, chẳng hạn chẩn đoán bệnh HIV-1, dùng phương pháp PCR chỉ mất một ngày so với 3-4 tuần nuôi cấy của phương pháp truyền thống. Thứ hai, đối với các tác nhân gây bệnh khó nuôi cấy hay không thể nuôi cấy thì kỹ thuật sinh học phân tử là giải pháp duy nhất, chẳng hạn Chlamydia, Brucella, viêm gan B,... Thứ ba, việc tạo dòng một gen dùng làm probe đơn giản hơn việc sản xuất kháng thể đặc hiệu, hơn nữa với một probe người ta có thể phát hiện tất cả các kiểu huyết thanh (serotype) của tác nhân gây bệnh, điều mà chỉ một kháng thể không thể làm được. Cuối cùng, nếu trước kia phải cần đến nhiều kỹ thuật (quan sát dưới kính hiển vi, nuôi cấy trên một loạt môi trường, miễn dịch học...) thì hướng chẩn đoán bằng sinh học phân tử chỉ cần một kỹ thuật (lai phân tử hoặc PCR).
Đối với tác nhân là virus, các kỹ thuật lai phân tử và PCR đã cho phép chẩn đoán nhiều nhóm như papillomavirus, HBV, HIV. Đặc biệt phương pháp lai tại chỗ còn cho phép xác định trực tiếp virus trên lát cắt sinh thiết.
Đối với các tác nhân vi khuẩn, đã có nhiều bộ kit chẩn đoán sinh học phân tử được thương mại hóa như: Mycobacterium tuberculosis, Mycobacterium pneumoniae,
Salmonella, Streptococcus pneumoniae, Haemophilus influenzae...
Đối với các tác nhân ký sinh trùng, người ta cũng đã thành công trong chẩn đoán Plasmodium, Schistosoma, Trypanosoma, Toxoplasma, Leishmania… bằng lai phân tử. Phương pháp PCR còn cho phép phát hiện tác nhân ngay trong vật trung gian truyền bệnh.
7. DNA fingerprinting
Sử dụng các trình tự DNA để nhận dạng một người, được gọi là DNA fingerprinting, là một công cụ rất mạnh để khảo sát tội phạm và các ứng dụng pháp lý
khác.
Trong một ứng dụng đặc trưng, DNA fingerprinting có thể được dùng để xác nhận một sự nghi ngờ hiện diện ở hiện trường gây án. Một mẫu DNA của máu, tóc, tinh dịch hoặc mô của cơ thể được thu thập từ hiện trường. Nếu mẫu là rất nhỏ, phương pháp PCR có thể được dùng để khuếch đại cho đủ DNA, sẵn sàng cho việc kiểm tra. Các mẫu DNA bổ sung được thu thập từ một hoặc nhiều nghi can khác.
Mỗi một mẫu được cắt với một hoặc nhiều RE, và kết quả các đoạn DNA được phân tách bằng điện di. Các đoạn trên gel được biến tính và chuyển lên màng nitrocellulose bằng thẩm tích Southern (blot). Một hoặc nhiều probe (được đánh dấu phóng xạ) sau đó được lai với màng nitrocellulose và được phát hiện bằng phóng xạ tự ghi. Kiểu của băng được tạo ra bởi DNA từ mẫu được thu thập ở hiện trường sau đó được so sánh với các kiểu được tạo ra bởi DNA từ những nghi can.
Các probe được dùng trong DNA fingerprinting phát hiện các vùng thay đổi cao của genome, vì thế cơ hội DNA từ hai người sản xuất chính xác cùng một kiểu băng là rất thấp. Khi một số probe được dùng trong phân tích, thì xác suất mà hai người có cùng một tập hợp các kiểu là gần như không có (trừ khi họ là cặp sinh đôi giống hệt nhau).
Mẫu khớp nhau từ hiện trường và nghi can có thể cung cấp bằng chúng rằng nghi can có mặt tại hiện trường vụ án.
Probe được dùng phổ biến nhất trong DNA fingerprinting bổ sung cho các trình tự lặp lại ngắn được tìm thấy rất nhiều trong genome người. Con người khác nhau rất nhiều về số lượng bản sao của các trình tự lặp lại này; vì vậy, các đa hình này được gọi là số lượng biến thiên của các đoạn lặp lại tuần tự (VNTRs).
DNA fingerprinting cũng được sử dụng để cung cấp thông tin về mối quan hệ huyết thống và các nguồn cơ thể sống khác. Ví dụ: DNA fingerprinting được dùng để xác định một số mẫu vi khuẩn gây bệnh than (anthrax) gửi bằng bưu điện đến những người khác trong năm 2001 tất cả là từ một nguồn mẫu.
8. Lập bản đồ gen
Một đóng góp có ý nghĩa của công nghệ DNA tái tổ hợp đó là cung cấp một lượng lớn các marker
(chỉ thị) di truyền được dùng trong lập bản đồ gen. Một nhóm trong số các marker được dùng trong lập bản đồ gen đó là restriction fragment length polymorphism (RFLP). RFLP là những biến đổi đa hình trong các kiểu của các đoạn DNA được tạo ra khi phân tử DNA được cắt cùng một enzyme hạn chế. Nếu DNA từ hai người được cắt bởi cùng một enzyme và kiểu của các đoạn DNA khác nhau được tạo ra, thì hai người này phải có sự khác nhau về các trình tự DNA của họ. Những sự khác nhau này được di truyền và có thể được dùng trong lập bản đồ, tương tự với phương thức mà trong đó sự khác nhau về allele được dùng để lập bản đồ các gen thông thường.
Theo truyền thống, bản đồ gen được dựa vào việc sử dụng các sai khác di truyền tạo ra những sai khác kiểu hình có thể quan sát dễ dàng. Đáng tiếc là do hầu hết các tính trạng bị ảnh hưởng bởi đa gen và môi trường, nên số lượng các tính trạng có cơ sở di truyền đơn giản thích hợp cho việc sử dụng bản đồ bị giới hạn. RFLP cung cấp một số lượng lớn các marker di truyền có thể được dùng để lập bản đồ gen của sinh vật.
Tài liệu tham khảo/đọc thêm
1. Dale JW and Von Schanzt M. 2002. From Gene to Genome. John Wiley & Sons, Ltd. West Sussex, UK.
2. Erlich HA. 1989. PCR Technology: Principles and Applications for DNA Amplification. Stockton Press, New York, USA.
3. Glick BR and Pasternak JJ. 2003. Molecular Biotechnology: Principles and Applications of Recombinant DNA. 3rd ed. ASM Press, USA.
4. Lewin B. 2000. Gene VII. Oxford University Press, Oxford, UK.
5. Maniatis T, Fritsch EF and Sambrook J. 1989. Molecular Cloning-A Laboratory Manual. Cold Spring Habor Laboratory, USA.
6. Primrose SB, Twyman R and Old RW. 2001. Principles of Gene Manipulation. 6th ed. Blackwell Science, Oxford, UK.
7. Rapley R and Walker JM. 1998. Molecular Biomethods Handbook. Humana Press Inc. New Jersey, USA.
8. Surzycki S. 2000. Basic Techniques in Molecular Biology. Springer-Verlag, Berlin, Heidelberg, Germany.
9. Walker JM and Rapley R. 2000. Molecular Biology and Biotechnology. Chapman & Hall Limited, London, UK.
Nguồn: Giáo trình Sinh học phân tử - Nguyễn Hoàng Lộc (chủ biên)